Dreaming of an Idea For Navigating Gene Sequencing IP A 2012 a podcast with Stephanie Murphy got our team thinking more deeply about gene patents and we spent a good year developing a technique which could potentially navigate many of them. Our bet turned out to be well placed as the technique we developed anticipated a particular Supreme Court ruling for it to be of value. In 2013, the SCOTUS decided that cDNA is patentable and that Natural DNA is not patent eligible. Many next generation sequencing techniques modify DNA substantially and are arguably more like cDNAs. The court has argued that to the extent the modification is indistinguishable from natural DNA it is not patent eligible. Thus DREAM PCR was published in Nature Biotechnology. It was submitted before the ruling. Our methods were also published in another open source journal for all to read for free. The link to this article can be found here!
Using DREAM for Decontamination Contamination from amplified products is a major concern when working with microbiological based techniques. Both culture and PCR amplify the pathogen for the purpose of detection and usually require sterile technique or clean rooms. PathoSEEK® is able to provide a contamination free technique through a proprietary process known as DREAM PCR which uses methylation specific restriction enzymes to prevent the carryover of amplified products from one reaction to the next. In the event that any DNA products from previous reactions are unintentionally introduced to subsequent reactions, an on board DREAM PCR™ enzyme will digest any DNA contaminants from previous tests. Any contaminant will have been tagged with a DREAM PCR™ methylation profile specifically targeted by the enzyme. The DREAM PCR™ enzyme is then deactivated so that the present reaction can proceed normally.
Link to our PLOS ONE Publication on DREAM PCR
Q&A: Courtagen’s Kevin McKernan Skirting Sequencing Gene Patents With Novel PCR Methods
*For the full story on this go to the GenomeWeb link below. They have great content and will keep you current in Genomics.
May 15, 2014
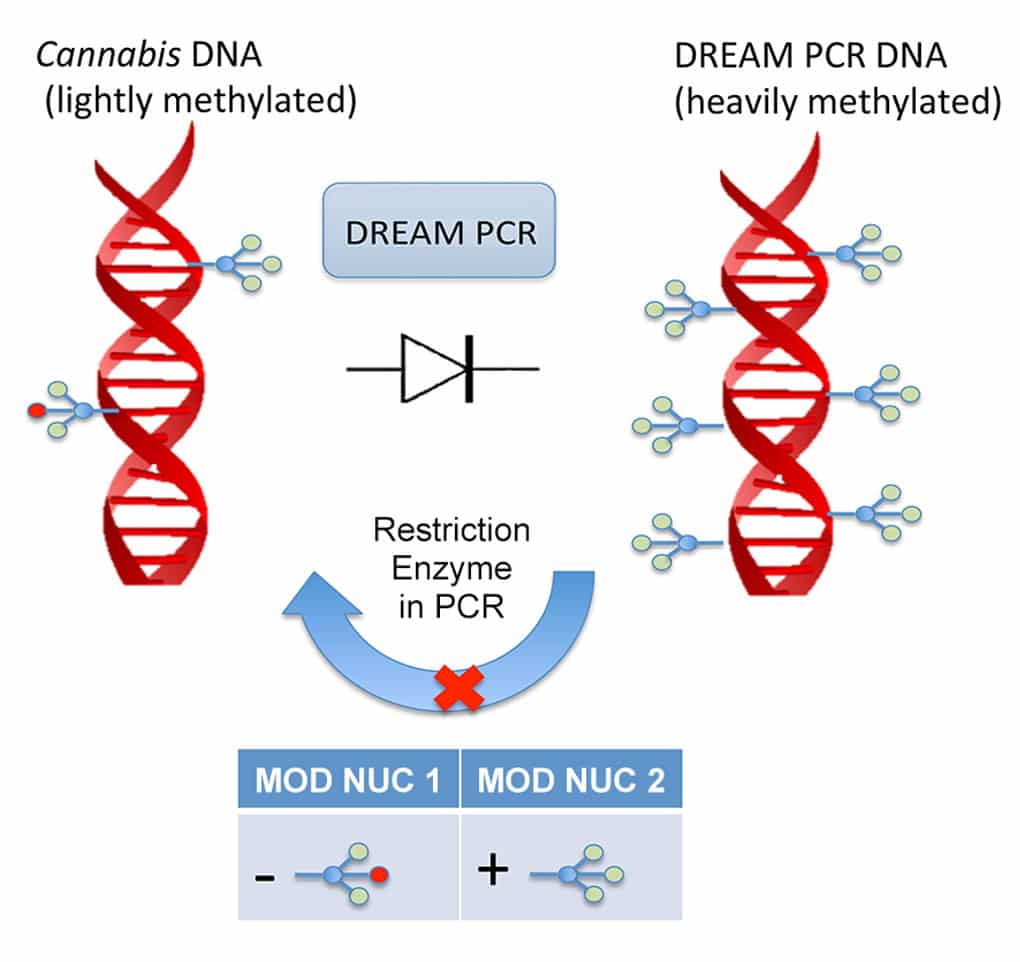
As CSO of Woburn, Mass.-based genetic testing firm Courtagen, Kevin McKernan recently co-authored a pair of papers with other Courtagen scientists describing novel PCR methods that he hopes will help others to avoid violating gene patents during clinical next-generation sequencing. McKernan believes that by hypermethylating DNA and using two methylation-specific restriction enzymes, researchers may be able to safely navigate the four main types of gene patents. The technique could also be used to prevent DNA from feeding back and contaminating early processing steps, as well to destroy patient DNA before it leaves a lab. McKernan spoke with PCR Insider this week about this work. Following is an edited version of the conversation.Your recent Nature Biotechnology paper reviews the impact of methylation on patents and property rights in genetics. You describe a PCR-based clinical sequencing strategy called DREAM that may challenge the interpretation of many patent claims. Could explain what DREAM stands for, and how it works? DREAM is an acronym that stands for decontamination-ready encoded amplification. The decontamination side of it is interesting. In clinical laboratories everyone is increasingly using these universal primers … [which] means that it’s very easy to falsely amplify [DNA from] a previous patient while you’re trying to amplify the next. What’s very interesting about the clinical sequencing space is that there is heavy regulation on how you handle patient data … under the HIPAA guidelines and the omnibus rulings, but there’s not as much scrutiny on the laboratory procedures in terms of … possibly let[ting] DNA leave your lab. People are not decontaminating their trash of DNA before it leaves, and so connecting any two bits of information — like simply amplifying something with Illumina primers, sequencing it, and seeing who was in that day — could be an identity hazard. We got interested in decontamination techniques, not necessarily due to concerns over people dumpster-diving our trash, but because we sequence samples that look for heteroplasmy, where we have to pick up sub-percentages of alleles. When you start getting down to those levels of genotyping, contamination begins to be a concern. You really need to make sure there’s no background DNA around. As we were developing this we began to see there are other issues that can persist here. We could have contamination on floors, on carpets, on pipettes, that could easily [allow] somebody … to universally amplify these PCR products and potentially sequence them and break patient confidentiality. That’s much harder to do on the data side right now than it is in the laboratory, and that made us think that we should get the laboratory procedures to be as encrypted and as safe as we have in the data front.
What exactly is DNA decontamination? It’s very analogous to what clinical labs have published in the past with the use of uracil, [which] you can target with a specific enzyme so you can eliminate the DNA after you’re done sequencing it. That’s the intent here. We switched that uracil base out with a methylated base. Uracil poses problems for a lot of polymerases. This is seen in bisulfite sequencing. It’s very difficult to get a polymerase to traverse the uracil. The polymerases are all much happier with the methylated bases and have much better incorporation kinetics. There have recently become available these methyl-specific enzymes that will only digest if the DNA is methylated. So that was really the swap that we did. It just so happens that in the course of amplifying DNA and reincorporating methylated bases, we believe we’ve traversed a lot of the gene patents that are out there. That was kind of a side effect we weren’t expecting out of the gate. How does that work? To be honest it didn’t work maybe a year ago, as of the SCOTUS ruling [in Association of Molecular Pathology et al. v. Myriad Genetics]. The Supreme Court ruled that any man-modification of DNA is patent eligible. To the extent that any man-modified DNA is indistinguishable from nature, then it’s not patentable. That has left people in a bit of a quandary as to what it really means. When you do next-generation sequencing, it’s very difficult to not man-modify the DNA. You’re going to put primers on the DNA, you’re going to target it, like doing exome sequencing. The example that was discussed in the Myriad case was the example of the cDNA. If you look at how cDNA libraries are made in that patent, it’s not very different from making an exome library. You are using probes to capture a subselection of the molecules, like you are with an exome capture. Then you’re somehow putting primers on them and amplifying them. Many people, I think, misread that ruling and jumped into Myriad’s space, and have since been brought to court … because the SCOTUS ruling really reinforced I’d say three out of four of the typical types of gene patents that are out there.
What are those four types? People will typically bucket gene patents into four categories. These are loose categorizations, but it’s the only way you can really begin to get your head around 4,000 gene patents, or 161,000 claims. Sliding oligo claim language is one of the four buckets. Arguably, the SCOTUS ruling annihilated those. That’s the one thing it probably did do, is that [it said] those aren’t specific enough. Composition of matter claims are often used for drugs. They are atomically interrogated, down to the actual atom. I had some experience with those when I was at Applied Biosystems, after they purchased Agencourt. The whole SOLiD versus Illumina patent estate fell down to whether sulfur was identical to nitrogen. And it turns out it was not, according to the court. In terms of DNA and composition of matter claims, if you put a methyl group on DNA it’s not the same thing. We can see that with PacBio sequencing, and we can see it in the fact that [Integrated DNA Technologies] demands that you specify whether you’re oligo is going to be methylated or not. We’ve also seen it in our papers. If we methylate the DNA, does it behave the same? We can sequence it on an Illumina sequencer, [but] we have to radically change the PCR conditions to make this work. So we feel really strongly that we’re not infringing on any composition of matter claim. The next set of claims is really a tricky set. A lot of lawyers have taken some creativity here, and say, “We claim any DNA sequence which encodes a polypeptide.” The reason they can do that is there is a wobble base, which means you get much more coverage on your claim language. For any given polypeptide sequence there is more than one DNA sequence which can encode it … So if you claim any DNA sequence which can encode a polypeptide sequence you are making claims to many more DNA sequences that are arguably off target to the gene. Now, it just so happens that Myriad used that exact language. Many others use that exact language. But in order for a DNA sequence to encode a polypeptide, it has to pass through translation and transcription. DNA which is heavily methylated is silenced in transcription. We’ve got a good argument that when we methylate DNA, you can’t encode anything because it stalls transcription. So, we think we’ve knocked out that section. The last category is another clever set of claims. They claim any DNA sequence which is complementary to another DNA sequence. What we’ve shown in the most recent paper is that, if you were to make an oligo of a given sequence like CATG over and over again, and you were to make the 3′ base of that change by a single base, you would see at most a 0.6 to a 0.7 degree change in Tm. If you were to add a methyl group to the C, you’ll see a 1.1 degree change. The reason that is important is that complementarity always comes back to Chargaff’s rules, A to T, C to G, and it has to do with Tm. Once you start methylating this, the Tm starts changing much more radically than it would if you had any other base composition change. We think we’ve got a strong argument that by hypermethylating [DNA] we’re radically changing the complementarity and therefore the claim language doesn’t stick. This [idea] has been reviewed by many lawyers. We decided to publish it because we wanted to have even more people argue whether it was right or wrong. Frankly, we don’t know the answer. We know that it’s a very complex space and with there being 161,000 claims, we’re not about to pay our attorneys to read them all. We’d rather have the public debate about it and see where things fall.
So that was the motivation for the Nature Biotechnology paper? Yeah, it was. We put it in the patent section … but we didn’t have enough space to go into the methods. We [also] wanted the methods to be open source, so we … put them inPLoS One, where we’d have enough page space to go through one by one, what you do in order to enable this.
For example, with the different enzymes —MspJI for 5mc, and AbaSI for 5hmc? That’s right, there are two different enzymes that are described in there. There’s also a lot of DMSO in order to get this to work. We mention it raises the Tm when you put that many methyl groups on a molecule. You can’t raise it to 110 [°C] to deal with it because you’ll just end up getting hydrolytic damage in the DNA. What you have to do is change the solvation through non-thermal methods. DMSO is great for that. We tested out all the way up to eight percent DMSO and showed what that curve looks like. Somewhere between three and four percent DMSO seems to be right for this library. The other new thing in the paper is the use of these two different enzymes. Many exome capture techniques that are out in the marketplace today require you to perform two amplifications. You amplify before you enrich, then you amplify after you enrich. You really need to have different bases at each of those steps if you want to ensure that one amplification isn’t going to contaminate the other. The first application we showed in Nature Biotechnology, we’re using [Agilent Technologies’] Haloplex, which is a different assay and only requires one amplification. So we can get away with just doing 5-methylcytosine. The moment we got into steps which were going from genomic DNA, which does have a little bit of methylation in it, then amplifying that, and then amplifying it again, we needed to start working with 5-hydroxymethylcytosine [as well]. There happens to be enzymes that discriminate the two that can create what we’ve termed a DNA diode, where DNA can only go unidirectionally through a lab. If it ever feeds its way back into other laboratory equipment, it is going to get digested by these enzymes that are very specific for digesting post-amplification products. Both of those enzymes are heat labile, so they’re in all of our reagents, and only when you heat them up, like in PCR, do you kill them. We have MspJI in our PCR cocktail, which means it’s constantly digesting any hypermethylated double-stranded DNA that’s around. The moment we kickstart PCR, it kills that enzyme and then that PCR reaction creates hypermethylated double-stranded products. If any of those find their way back into the previous step, they’re chewed by the enzyme. The same thing is true with the second amplification. We have hydroxymethylated bases in there, which are slightly different than methylated C, and AbaSI will specifically digest hydroxymethlyated C without digesting methylated C. That’s important because the input DNA at that point is going to be hypermethylated from the first PCR. So we needed to have an enzyme that could differentiate those two. We think there’s a lot of applications for this outside of just these assays, and I think if you ever want to make something portable, or [develop] field amplification techniques, where you don’t want to worry about clean rooms and all the hyper-cleanliness, or disposable tips and robots, there’s all types of areas where this will simplify the PCR process because of the built in error correction.
Are there any potential pitfalls? Genomic DNA is going to be lightly methylated. So, if you apply MspJI there, you’re arguably going to deplete your first exons, and your CpG islands will get digested. Some people have asked if that’s a problem for your capture. If you’re running around cutting all the regions that are methylated they’re not going to be in your exome to capture. It turns out that doesn’t appear to be the case with the Haloplex protocol [we use] because it turns everything into a single-stranded circle at the point at which we’re using this enzyme. That’s not to say it wouldn’t create problems for other assays, people just need to be aware of that. On the flip side, about 0.1 percent of the DNA is hydroxymethylated, so you can always switch to that enzyme and rearrange what we’ve done if people are concerned about losing the first exons or the CpG islands in their assay.
Déjà vu PCR is the method using both enzymes. You published this as a method and it sounds like you want people to try it, and hopefully publish on it also. Yes. There are also a couple companies that have asked us to license it to put it into kits. So, it will probably come out in the form of a kit, and what they call it is up to them. Déjà vu is really meant to emphasize we’re doing it again. It’s methylated one time and then remethylated with a slightly different methyl group the second time. [This would likely be] a kit for keeping PCR clean. I don’t think anyone is going to turn this into a spray for the lab trash. I have a feeling it would be in everyone’s PCR tools. This is important for capturing clean sequences out of mitochondrial DNA, mosaicism, or heterogeneity in cancer. You have to call five percent allele frequencies, and you’re questioning, “Is that a contamination event, or is that actually a rare population of tumor cells that I need to worry about?” You can waste a ton of time looking at contamination questions. It’s better just to build tools that safeguard against it.
You wrote in your Nature Biotechnology paper that you believe gene patents are immoral. Would you like to expound on that? I spent four of five years of my life burning myself an ulcer at the Human Genome Project to make sure that the genome was public and that there were Bermuda principles involved. The group that we vilified in that process was Celera Genomics because they were going to patent some genes. When all was said and done, the National Institutes of Health has more gene patents than Celera. That has not followed the Bermuda accords by my standards. That has gotten somehow into a hypocritical morass of: What was the genome project for? To me, putting all that effort in, I expected when we went to sequence sick kids in 2014, we wouldn’t be threatened with lawsuits for doing it. Now we’re in a scenario where all these start-up companies are coming to play, to bring next-generation sequencing to the market and potentially help a tremendous number of sick people, and 20 percent of genes are thought to be patented. That’s not fostering innovation. If that’s what the patent system is designed to do, it is failing horribly.
So in terms of your PCR-based method, it may allow other people to get around this problem? We hope so. We can’t say that with 100 percent certainty because we haven’t gone and read 161,000 gene patent claims. I wouldn’t recommend anyone go do that. That’s just a tremendous amount of money wasted that should be put toward building better assays and sequencing more patients. [Our method] is hopefully something that can get sold and spread around to the world. It’s been published and there are companies that are talking about commercializing it. We’re not in the best position to commercialize it because we are in fact a clinical sequencing lab. For us to build kits to do this we would need a partner, and … the intent of some of these agreements is to get leverage through a supply chain and sales channel that is much more powerful than ours. That should enable other labs to potentially adopt this and not have to spend their time with these sorts of [legal] questions.
Your company launched two next-generation sequencing-based tests last week for neurodevelopmental disorders, developmental delay, intellectual disability, and autism spectrum disorder. Do these use your new methods as well? All of our methods use the Haloplex plus the DREAM PCR for the panels. We even have that working in whole exomes as well. It’s been applied to everything we do.
Are there any other interesting current projects? The other assay that we are working on, and hope to have launched at the end of the year, is to move the whole chromosomal microarray problem into a next-gen sequencing assay. That’s typically the front line of defense. People start with a chromosomal microarray to look for a megabase-sized copy number variation. There’s no reason that should be done in a chip anymore. There are assays that would move that on to a next-gen platform that have already been published and are fairly easy to implement. [For example], Zheng et al. published the replacement of an Affymetrix 1.8M SNP array with RAD-Seq on a SOLiD.
Articles
