
Cannabis testing labs are being presented with a wide variety of microbial testing solutions, each claiming they can shorten test times, increase sensitivity, and lower costs. Given microbial testing is currently the bottleneck in lab turnaround time, products claiming faster results are particularly tempting. However, labs should be wary.
We understand enrichment is a tempting corner to cut, especially considering we have observed enrichment bias when studying culture-based enrichment (McKernan et al. 2015; McKernan et al. 2016; McKernan 2018). However, the AOAC has set the SMPR for cannabis microbial testing to require enrichment. Any test that cuts this corner will not be compliant with the AOAC SMPR.
The primary reason we, and presumably AOAC, require enrichment as part of our microbial safety testing protocol is to overcome the sampling bias. Testing methods typically use 10-15ml to homogenize 1 gram of cannabis, and not all of this culture is placed onto a petri dish or into a PCR reaction. Enrichment must be performed to overcome this sampling bias. For example, if only 200µL of 10ml is placed onto a petri dish, only 1/50th of the homogenate is being sampled and tested. If only 1 CFU is detected you can easily multiply this by 50 to get the original CFU estimate, but if zero CFUs are detected, you don’t know if you had zero or 49 CFUs. States are asking for single CFU sensitivity in 1 gram samples and it can’t be known for certain unless you enrich the sample to compensate for this sub-sampling bias.
The enrichment step allows for at least a 50-fold increase in microbial content. The doubling times of microbes vary and must be considered when enriching for this subsampling. We acknowledge that culture-based enrichment creates a bias for microbes that grow efficiently in the enrichment broth, but a bias is better than guaranteed false negatives that will occur due to sampling bias.
Centrifugation ≠ enrichment
Methods have been proposed to cut the enrichment corner via centrifugation. We tested this method and found it is flawed.
Proposed Protocol to circumvent enrichment with centrifugation (Taken from vendor’s public YouTube tutorial):
- Centrifuge and decant supernatant from samples (physical enrichment)
- Centrifuge at 50 x g for 3 min
- Transfer supernatant to a clean microcentrifuge tube
- Centrifuge at 14,000 x g for 3 minutes
- Discard supernatant
- Resuspend the microbial pellet in the biochemical enrichment agent (biochemical enrichment)
- Incubate at 37°C for 20 minutes
- Transfer supernatant to a clean microcentrifuge tube
- Centrifuge at 14, 000 x g for 3 min
- Discard supernatant
- Perform a sample preparation as stated in SOP
- Add 35 µL Sample Digestion Buffer, vortex and quick spin
- Heat at 95°C for 10 minutes
- Add 5 µL Neutralization Buffer, vortex and quick spin
- Add 25 µL Sample Preparation Buffer, vortex and quick spin
- Heat at 55°C for 45 minutes, vortex and quick spin
- Heat at 95°C for 15 minutes, vortex and quick spin
- Samples ready for PCR
This proposed process is scientifically flawed for the following reasons. The first step of this protocol (“physical enrichment”) asks for a 50 x g spin for 3 minutes where the supernatant is then carried on to the next step and the pellet discarded. This first spin is believed to only remove large debris from the homogenization process, but given the endophytic nature of many microbes, this assumption is one that must be tested.
The first question to ask is how much live versus dead DNA is in the pellet of this first 50 x g step as this pellet is discarded from analysis in the suggested protocol. Any detection of live DNA in this pellet implies microbes are being missed and the test will under detect patient risk.
The sedimentation rates of E. coli and DNA are well studied and it is well known that live E.coli can sediment at 1 x g on your benchtop. This is why growing E. coli overnight often results in a visible pellet of cells at the bottom of your tube the next morning. This segregation of cells versus free floating chromosomes is even harder to accomplish with filamentous fungi known to clump and known to make 1-5um sized spores.
Putting centrifugation to the test
To assess the live and dead DNA in this pellet we grew E. coli to stationary phase at 37°C. Stationary phase will contain both live and dead E. coli thus mimicking a cured or partially sterilized cannabis flower. This sample was diluted to 5% vol/vol in TSB (200µL /4mls). We applied 42 X g centrifugation (less than 50 X g) and used qPCR with and without our Grim Reefer® Free DNA Removal Kit (salt tolerant nuclease) to determine the ratio of live and dead DNA in the pellet and in the supernatant.
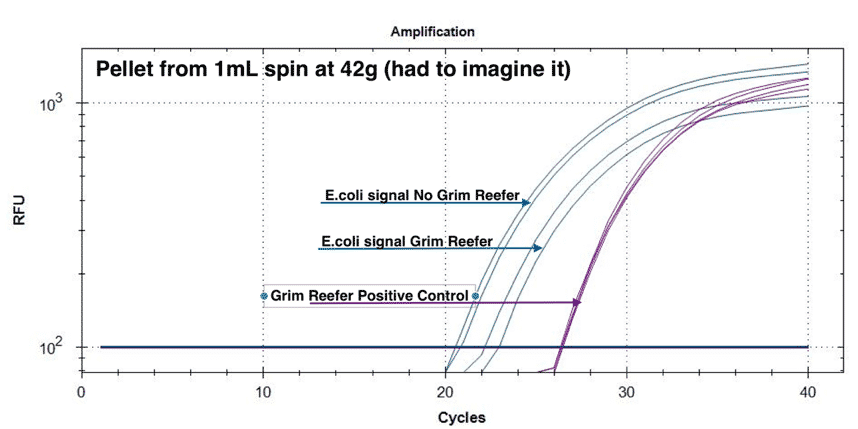
If a lab uses this “physical enrichment” to skip enrichment and is only harvesting the supernatant of this step for microbial analysis, they are leaving behind a significant portion of the pathogenic load in the pellet. This will fail to detect pathogen E. coli as the majority will have been spun out of the sample in the first 50 X g step. It is important to note that we used even lower g forces for this study to give the authors the benefit of the doubt.
A lower failure rate ≠ safer cannabis
This may be advantageous for microbial kit provider as their assay will appear to have a lower failure rate and attract more growers to lobby for their kit’s use. We see this happen in states that allow plating and molecular methods. The growers lobby for the test they can counterfeit with microwave or other sterilization treatments as they fear PCR will penalize them for dead DNA. In the end, the labs will be ring tested with standards which will highlight these sensitivity deficits.
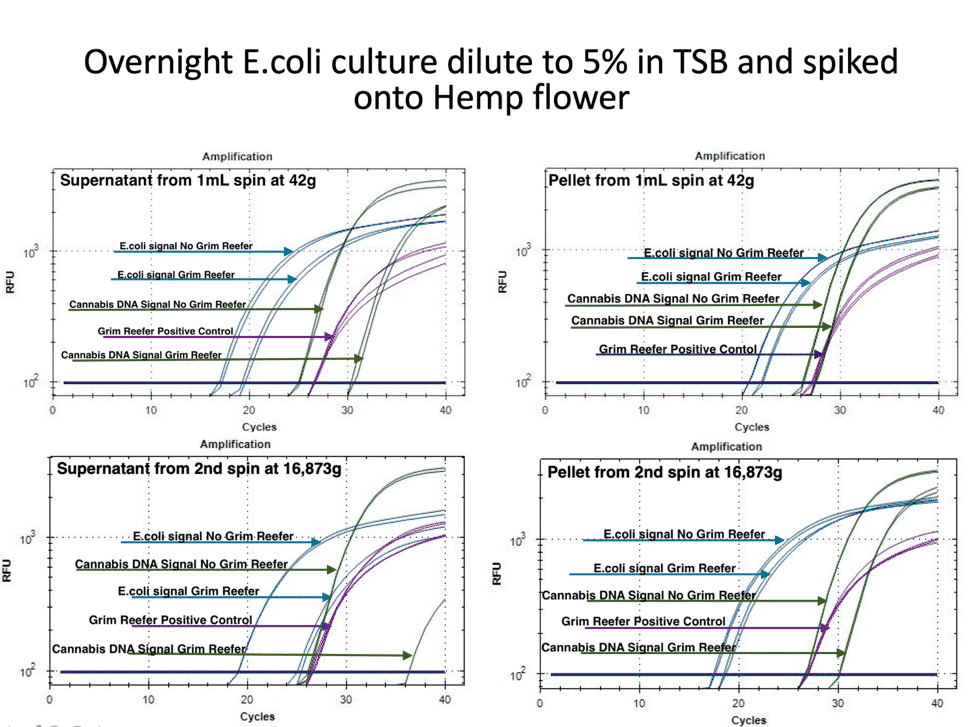
To expand on this study, we repeated this in the presence of Hemp matrix with an internal Cannabis DNA control. The internal Cannabis DNA controls is critical to confirm you are lysing open plant cells and accessing the endophytic fraction of the plant. Many microbes are Cannabis endophytes and controls must be in place that confirm the lysing conditions are accessing these endophytes. Simple spike-in DNA controls do not provide this important control.
Figure 3 demonstrates a similar loss of pathogenic risk in the first centrifugation step. The pellet from the 1ml spin at 42 x g demonstrates both live and dead DNA are captured in the pellet and this DNA is removed from the microbial test. The cannabis DNA in the pellet is mostly live DNA, presumably from the trichomes often found in these pellets. The supernatant of this pellet still has live and dead DNA in it and the plant DNA has a larger amount of dead DNA.
In this experiment we also quantified the DNA before and after the second centrifugation step. The pellet of this 14,000 rpm (16,873 g) step still has live and dead DNA present while the supernatant is predominantly dead DNA for both E. coli and hemp. To give the authors the benefit of the doubt we spun this sample harder than their recommended 14,000 x g.
Conclusions
This data clearly demonstrates that “physical enrichment” removes pathogenic microbes from the sample being tested (remains in the discarded 42 x g pellet) and is a risk to cannabis patients. It is important to note that E. coli DNA is circular and usually less than 5Mb in size. Fungal genomes can be as large as 200Mb and contain mitochondrial DNA that is often smaller than E. coli, making them more challenging than E. coli to differentially sediment from live cells with centrifugation. Labs can readily confirm this result in their own lab in an afternoon. Grim Reefer® reagents and microbial qPCR reagents are available on our web store. If your competitor is taking this short cut to compete on turn-around time, show this data to your local regulator so they can understand the risk this presents to immunocompromised patients, and to you and your business.
In conclusion, given the AOAC demands for enrichment-based methods in the SMPR and evidence of these short cut techniques reducing the microbial count of a given test, side stepping enrichment is too risky. The current method is flawed and produces results no one can trust.
References
McKernan K. 2018. Microbiological examination of nonsterile Cannabis products: Molecular Microbial Enumeration Tests and the limitation of Colony Forming Units. OSF doi:10.31219/osf.io/vpxe5.
McKernan K, Spangler J, Helbert Y, Lynch RC, Devitt-Lee A, Zhang L, Orphe W, Warner J, Foss T, Hudalla CJ et al. 2016. Metagenomic analysis of medicinal Cannabis samples; pathogenic bacteria, toxigenic fungi, and beneficial microbes grow in culture-based yeast and mold tests. F1000Research 5: 2471.
McKernan K, Spangler J, Zhang L, Tadigotla V, Helbert Y, Foss T, Smith D. 2015. Cannabis microbiome sequencing reveals several mycotoxic fungi native to dispensary grade Cannabis flowers. F1000Research 4: 1422.







